Биоинформатика
Прочтение (рид) - это
последовательность, полученная из секвенатора
Cкаффолды
Удлиненные контиги (за счет дополнительной информации)
набор контигов, про который известно, что контиги идут в данном порядке; разрывы между контигами обычно заполняются буквами N в числе, равном предполагаемой длине разрыва
Для того, чтобы автоматически выбирать и фильтровать прочтения по качеству используют программу
Trimmiomatic
FastQC
первый и самый важный этап обработки данных
написана на Java
Выдает результат в формате .html
позволяет оценить риды по ряду параметров
Для каждой позиции выдается ящик с усами (boxplot) для качества
Прямые праймеры отжигаются
5' -> 3'
цепь 5'______ 3'
3'______ 5'
Trimmomatic
это быстрый многопоточный инструмент командной строки, который можно использовать для обрезки данных Illumina (FASTQ), а также для удаления адаптеров
Триммоматик используется для
для обрезки адаптеров после секвенирования
Принцип работы SPAdes
ищет эйлеров путь, который будет контигом (граф Де Брейна в котором в вершинах записаны k-меры, а на ребрах - их перекрытия K-1меры)
Средняя длина контига
2000 пн
Метод скользящего окна
- берет массив данных и расчитывает среднее для опр. количества чисел (3) [2357] - 3.3, 5 и т.д.
Trimmomatic использует скоьзящее окно чтобы обрезать последовательность, когда среднее качество нуклеотидов становится <20 (или любое другое Q).
Гаметонов путь
позволяет найти путь в связном графе, чтобы он захватывал все вершины только 1 раз. Нет алгоритма для его нахождения, можно только перебором
Эйлеров путь
проходит по всем ребрам 1 раз
Гамильтонов путь
путь, проходящий по всем вершинам графа и притом только по одному разу
Алгоритм Хиерхольцера
позволяет найти Эйлеров граф (ищем место, в котором ошибка, начинаем с него)
OLS алгоритм
использовался при секвенировании небольших геномов по методу Сенгера (большие риды). Получаем риды, ищем перекрывающиеся участки (каждого с каждым), объединяем в одну последовательность и получаем кусочки генома большей длины – контиги.
граф Де Брейна
ориентированный граф, в котором ребро соединяет такие вершины, между которыми есть перекрытие длиной l
Глубина прочтений
сколько ридов содержит конкретный нуклеотид при сборке генома
Проблемы секвенирования:
повторы (одинаковые кусочки трудно соединить), композиция генома и неравномерность покрытия (нодопредставленность гетерохроматина)
Ширина покрытия (при секвенировании)
какая часть молекулы покрыта прочтениями (если читаем вслепую - неизвестна)
статистики длин набора контигов для оценки качества сборки генома (меры качества сборки)
N50 (длина самого короткого контига в наборе контигов, упорядоченных по длине, покрывающих половину сборки) и L50 (номер этого контига)
L50 —
минимальное число контигов, чья суммарная длина не менее половины суммарной длины сборки
SAM формат это
- sequence alighment map - файл, где хранится карта выравнивание ридов на референс
BAM формат это
(Binary Alignment Map) - файл SAM переведенный в бинарный вид (т.к. он очень большой)
в SAM-файле
хранится карта выравнивания ридов на референс.
Количество укорененных и неукорененных филоген. деревьев (формула)
неукорененных - (2n-5)!!
двойной факториал - произведение всех нечетных чисел до этого числа.
Количество укорененных филоген. деревьев - (2n-3)!!
качество сигнала Q характеризуется (какое значение, формула)
значением Phred с вероятностью ошибки p
Q = -10log(p)
Типы аннотаций (3)
Основанная на гомологии - структурная
Основанная на свойствах генов (ab initio) - функциональная
Основанная на экспрессии генов (экспериментальная)
Аннотация, основанная на гомологии (что использует)
Использует гены модельных объектов + статистические данные
Основывается на выравниваниях
Точная, позволяет получить позиционную и структурную информацию
Аннотация, основанная на свойствах генов (особенности)
Выдает стабильные предсказуемые результаты для большинства организмов
Неточная, не позволяет присвоить функциональную информацию
Большое количество недопредсказаний/ложных предсказаний
Статистическое предсказание генов (что позволяет их найти)
Различные частоты кодонов (codon usage bias)
ГЦ-состав
Старт и стоп-кодоны
Сайты сплайсинга
Транскрипционные и трансляционные сигналы
Формат GFF
(genome feature format) - хранит данные об аннотации
Для проведения сборки используется программа
Spades
Для картирования прочтений используется программа (в нашем случае)
bowtie
Секвенирование второго поколения
фрагментация, лигирование адаптеров, амплификация фрагментов для усиления сигнала
Секвенирование по Сэнгеру (+-)
Риды 400-900 нуклеотидов
высокая точность
Не позволяет секвенировать большие последовательности (долго и дорого)
454 пирофосфатное секвенирование
PPi реагирует с фденозитфосфосульфатом с помощью сульфурилазы. образуется АTP, который использует люцифераза для создания свечения.
Проблемы сборки ридов:
Повторы
Гетерозиготность
Полиплоидия
Контаминация
Ошибки секвенирования
Неравномерность распределения ридов (из-за GC-состава, например)
Последовательность получения генома из секвенирования
Получение ридов
Контроль качества
Тримминг и фильтрация
Контроль качества
Сборка генома
Методы множественного выравнивания (программы)
MUSCLE (итеративно-переборный), ProbCons (точный, но затратный), MAFFT (очень быстрый, работает с FFT от последовательностей), ClustalW(классическое прогрессивное выравнивание, взвешивает последовательности на
основе их guide tree)
Псевдогены
Нефункциональные аналоги структурных генов
Аннотации хранятся в файлах форматов
.gtf, .gff3
Последовательность анализа транскриптомных данных
Получение ридов
Контроль качества
Тримминг и фильтрация
Выравнивание / псевдовыравнивание
Подсчет экспрессий (анализ)
Последовательность действий при работе с секвенированием РНК
Получение ридов
Проверка качества
Выравнивание
Анализ
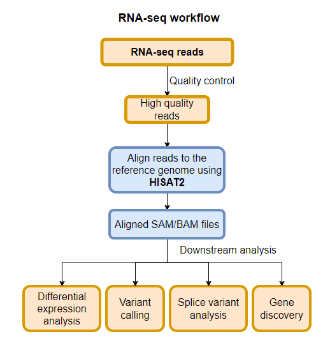
Проблемы секвенирования РНК:
Транскриптов больше чем генов
У транскриптов разная представленность
Надо избавляться от рибосомных РНК
Нестабильна
Картирование коротких прочтений
метод анализа результатов секвенирования 2 пок., состоящий в определении позиций в референсном геноме или транскриптоме, откуда с наибольшей вероятностью могло быть получено каждое конкретное короткое прочтение. Обычно является первой стадией в обработке данных в случае, если известен геном
Проблемы картирования:
Вариабельность генома
Ошибки секвенирования
Повторы
Время / большие объемы данных
Пакет samtools используется для
обработки BAM-файлов (сортировки по координате вхождения в референс, удаления дубликатов, фильтрации по количеству вхождений)
IGV
(Interactive Genomic Viewer) - при загрузке туда референса и ВАМ-файла позволяет визуализировать картирование
Основа алгоритмов множественного выравнивания
Прогрессивный подход
p-value
вероятность получить выравнивание, полученной по той же базе данных с таким же запросом, с таким же или большим весом по случайным причинам
Если оно большое - нулевая гипотеза верна
E-value
математическое ожидание того, что эта последовательность (результат BLAST) нашлась случайно. Чем она ниже, тем лучше.
Gblocks
компьютерная программа (написанная на языке ANSI C), которая устраняет плохо выровненные позиции и расходящиеся участки выравнивания последовательностей ДНК или белков. Используется (нами) для маскирования мусорных позиций
dN/dS отражает
скорость накопления мутаций
селективное давление
dS отражает
скорость синонимичных мутаций
dN отражает
скорость несинонимичных мутаций (изменение амк)
чему равно dN/dS при отсутствии эволюционного отбора
В отсутствие эволюционного давления синонимичная и несинонимичная скорости равны, поэтому соотношение dN/dS равно 1
dN/dS при движущем отборе
несинонимичные мутации полезны. N>S
коэффициент замены аминокислот благоприятствует отбору, и dN/dS > 1
dN/dS при стабилизирующем отборе
<1
N<1
Eстественный отбор предотвращает замену аминокислот, поэтому dN будет ниже, чем dS, и dN/dS < 1.
Ортологи -
гомологи, разделение которых произошло в результате видообразования
Паралоги -
гомологи, разделение которых произошло в результате дупликации.
bootstrap - нужен для
анализа качества построенных деревьев
IQtree -
– инструмент для построения филогенетических деревьев, оценки моделей эволюции и оценки достоверности полученных деревьев. IQ-TREE принимает в качестве входных данных множественное выравнивание последовательностей и реконструирует эволюционное дерево, которое лучше всего объясняется входными данными.
IQtree использовалась в нашем случае для:
Оценки модели нуклеотидных/аминокислотных замен
Построения филогенетического дерева
Статистической оценки полученного дерева (bootstrap)
